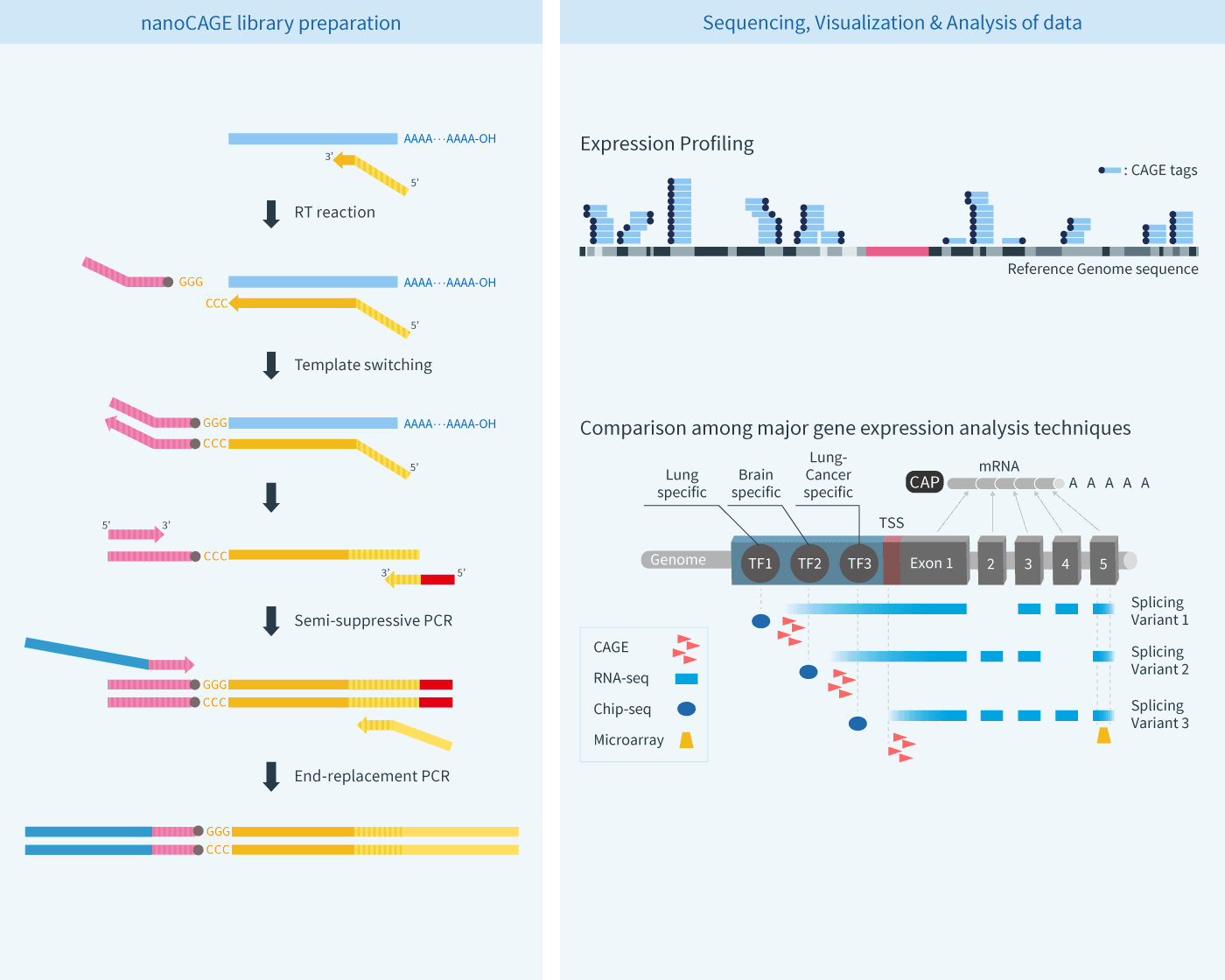
nanoCAGE™ Library
nanoCAGE/CAGEscan is a modified technique of CAGE that enables genome-wide promoter analysis of small-amount of samples. Utilizing a template-switching reaction and a semi-suppressive PCR, amount of sample required is significantly reduced. CAGEscan basically sequence and map short tags derived from the 3’-end of cDNA (5’-end of mRNA), thereby quantifying the frequency of tag sequences. Like CAGE, nanoCAGE/CAGEscan can accurately identify promoter sites for each transcript and obtain expression profile of respective promoters in genome-wide fashion. Several articles performing transcriptome analysis with nanoCAGE in human and mouse have been published.
With a sensitivity one thousand times higher than CAGE, nanoCAGE/CAGEscan presents powerful new possibilities for the analysis of samples with an amount as little as 50 ng of total RNA. NanoCAGE/CAGEscan overcomes the difficulty on obtaining large quantity of cells for canonical CAGE analysis, supplying a possibility for diagnosis of diseases such as detection of cancer and neuron disorder in future.
As the joint developer with RIKEN, DNAFORM library preparation service provides the end-to-end solution for gemone-wide semi-quantitative expression analysis including library construction, high- throughput sequencing with the Illumina HiSeq platforms and data analysis.
Applications
- Genome-wide gene expression analysis with tiny samples
- Prediction of promoter sites with small amount of sample
What can nanoCAGE/CAGEscan do
Overcoming the sample amount requirements
Only 50 ng of total RNA is required.
Unique data
CAGE enables experimental identification of transcriptional start sites and quantification of promoter activity.
Wide dynamic range
CAGE also enables detection of rare transcripts.
Learn more about:
How to choose CAGE and nanoCAGE/CAGEscan
| Library | Advantage | Disadvantage |
| CAGE | Quantitative expression profile without PCR bias. More cap-dependent compared to nanoCAGE. | 3μg of total RNA is required (If possible, please provide 12μg of total RNA for PCR free CAGE library preparation service). |
|---|---|---|
| nanoCAGE/CAGEscan | Only 50 ng of total RNA is required (100ng is recommended). 3'-end analysis with the same library is possible |
PCR step during the library preparation may cause PCR biases on the expression profile. |